
- Background
MADNESS does quantum chemistry
- DFT functionals for the user
A molecular wave function may be determined using the ground state density only. For all but the simplest local density approximation functionals we use the density functional library libxc. The user may request a functional according to the general syntax
where FUNC1 etc are the name of a libxc functional, and the weight describes the admixture coefficient
or use the following predefined functionals (not case sensitive)
As an example, the PBE0 functional would be expressed in the general syntax as
Expert parameters are RHOTOL, RHOMIN, and GGATOL, which determine at which density value threshold (RHOTOL) the density will be set to RHOMIN inside libxc, and at which density the gga potential will be munged, since it might not be bound.
- DFT functionals for the developer
A DFT functional is most easily constructed through the use of the madness::XCOperator class in SCFOperators.h
where the arguments to the constructor are the world and a pointer to a madness::SCF or madness::Nemo calculation. The constructor will create the necessary density and density gradients (in XCOperator::prep_xc_args() ). Note that the ispin argument (0 for alpha, 1 for beta) is only important for the computation of the xc potential, and may be changed later on to avoid re-creation of the density (gradients). The functional information is passed to the constructor through the calculation parameters.
A DFT energy and potential can be constructed as
Direct application of the XC functional to a vector of orbitals mo may be accomplished as
If a response kernel is requested (e.g. in CPHF equations, or in linear response), the kernel itself is numerically unstable. To some extent this instability is circumvented by performing as many operations as possible on a fixed grid, so that only the result of the kernel application is available
- Internal implementation details
For LDA functionals the density is processed as-is, only very small and negative densities are set to zero (through the RHOTOL and RHOMIN arguments).
For GGA functionals the reduced density gradients 
![\[
\rho = exp(\zeta) \\
\nabla \rho = \rho \nabla \zeta \\
\sigma = |\nabla\rho|^2 = |\nabla \zeta|^2 \rho^2 = \chi \rho^2
\]](form_34_dark.png)
which defines the quantities 



Response kernels for GGA are even more numerically unstable than the XC potential. We use the same log-derivative trick as before, which fixes the long-range noise. However the intermediate potentials have to be multiplied with the gradients of the (perturbed) density, followed by taking the divergence, which is equivalent to applying the Laplacian to the (perturbed) density. Numerical noise is amplified and convergence during the SCF or CPHF iterations might not be achieved. Furthermore, the perturbed density is not strictly positive, so its logarithm is not defined. We can pull an unperturbed density out of the perturbed density gradient to regain some stability
![\[
\sigma_\mathrm{pt}=\nabla\rho\cdot\nabla\rho_\mathrm{pt} = \rho\left(\nabla\zeta\cdot\nabla\rho_\mathrm{pt}\right)
\]](form_39_dark.png)
The code in XCOperator works as follows
- compute
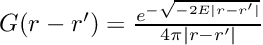
- call xc_potential or xc_kernel_apply
- call
madness::XCfunctional::make_libxc_argsto munge the density and density gradients – convert spin density to full density if spin-restricted - call libxc functions to compute the functional derivatives of the density functional
- multiply intermediate quantities to the functional derivatives
- return scalar intermediates, for the exact description see in madness::XCfunctional::vxc and madness::XCfunctional::fxc_apply
- call
- multiply the semilocal intermediates with the density gradients, take div operator